

- Bone Health
- Immunology
- Hematology
- Respiratory
- Dermatology
- Diabetes
- Gastroenterology
- Neurology
- Oncology
- Ophthalmology
- Rare Disease
- Rheumatology
BioRationality: A Dr Sarfaraz Niazi Column—FDA Discloses Changes to Drug Review Process, Biosimilar Licensing
Sarfaraz K. Niazi, PhD, explains the FDA's changes to the drug review process, the significance of each change, and the published pieces of work that helped inform the FDA's decisions.
The FDA has always been proactive in bringing science ahead of archaic regulatory beliefs; one example is the FDA's Division of Applied Regulatory Science (DARS), which conducts multidisciplinary studies to answer scientific questions and solve regulatory challenges, leading to modifications in the regulatory guidelines. Recently, the DARS' 21 top scientists published a pivotal paper summarizing some of their recent achievements based on the plans for 2021, many of which will impact the development of biosimilars.[1] I had the privilege of serving as a reviewer of this paper.
The capabilities within the DARS include laboratory-based research specializing in omics, bioanalysis, micro-physiological and cellular systems, immunology, electrophysiology, and silico research performed by informatics and computational modeling groups. In addition, DARS conducts clinical research focused on facilitating new and biosimilar/generic drug development and assessing the safety of marketed drugs.
Finally, using applied research, DARS investigates questions related to clinical pharmacology, medical toxicology, systems pharmacology, chemistry, and biology.
The following projects were discussed in the paper:
- Emergent regulatory and public health questions: sunscreen active ingredient absorption, over the counter heartburn medication ranitidine, adverse events of COVID-19 therapeutics, optimizing COVID-19 therapies, addressing the opioid epidemic, optimizing opioid reversal agents, and opioid drug-drug interaction effects on respiration; Public Health Assessment via Structural Evaluation
- Better predicting potential drug safety issues: predicting immune-mediated adverse events, leveraging molecular target information to predict safety issues, predicting drug interactions with (quantitative) structure-activity relationship [(Q) SAR] models, and drug-drug interaction studies
- Facilitating biosimilar and complex generic development: pharmacodynamic biomarkers for biosimilar development and approval, novel methods to assess immunogenicity for biosimilars and complex generic drugs, supporting generic drug development in pediatrics, and investigating novel approaches to establish bioequivalence for complex generic products
- Advanced therapies for rare diseases: drug approvals using nonclinical data, assessing the potential for induced pluripotent stem cells to streamline drug development for rare diseases, and identifying molecular targets for pediatric cancer
- New alternative methods: new alternative methods to animal testing, implementing regulatory science, from applied research to leading international regulatory guideline updates
- Liming drug impurities and extractable/leachable compounds: (Q)SAR assessment for drug impurities, structure-activity assessments for extractable/leachable compounds, (Q) SAR evaluation of nitrosamine impurities to assess carcinogenic potency, drug overdose labeling, and new models for measuring the emergence of antimicrobial resistance
- Resulting regulatory impact: supporting regulatory action: drug labeling change, affecting regulatory policy, and drug development tools
Expediting Biosimilar Development
Bringing more drug competition to the market and addressing the high cost of medicines is a priority for the FDA and the HHS, an effort exemplified by the Drug Competition Action Plan in2017 and the Biosimilars Action Plan in 2018. These plans outlined concrete steps the FDA would take to remove barriers to biosimilars and generic drug development, including prioritizing biosimilar applied research projects.
Immunogenicity of Biosimilars
A factor influencing the rapid development of biosimilars is the ability to predict the potential risk of a stronger immune response (immunogenicity) in humans to a proposed biosimilar than that seen with the innovator product.
For most biosimilars, this is still assessed through clinical trials. DARS is studying the ability of nonclinical approaches to predict immunogenicity risk without conducting a clinical trial. This includes assessing specific in vitro assays and cell types, in vivo models, and identifying valuable controls. These methods have the potential to identify products with immunogenicity risk sooner and streamline the development of biosimilars and certain complex generic drugs.
DARS compares theabilityofdifferent nonclinical models to predict immune-mediated adverseeventsfrombiologicaldrugproducts. For example, DARS has testedtheuse of novel nonclinical models to predict cytokine release syndrome, a potentially life-threatening complication associated with biological products, and showed that nonclinical models could effectively demonstrate this adverse event.[2],[3]
Additionally, after successfully demonstrating immune-mediated activation in a nonclinical model, investigations continue for checkpoint inhibitor oncology treatments for which adverse events cannot be predicted using computational, in vitro, or conventional nonclinical methods.[4]
Since the (PHASE) can be used to predict the activity of new proteins and identify their immunogenicity, in silico models that demonstrate similarity should also show similar activity and immunogenicity. For example, the 3-dimensional (3D) structure of proteins depends entirely on the primary amino acid sequence. So, if a biosimilar candidate has the same sequence, why should its 3D structure differ from the reference product? Since the 3D structure is entirely responsible for activity (receptor binding) and immunogenicity, why should a biosimilar candidate not be the same and require additional testing?[5]
Molecular Target
DARS significantly focuses on leveraging molecular target information to predict safety issues. Knowing a drug’s molecular targets can identify a drug’s effects and potential safety concerns early. DARS leads multiple efforts collating information about a drug’s known and predicted targets to identify potential safety concerns.
For example, DARS developed multiple computational methodologies, including machine learning, to predict a drug’s adverse effects based on the biological receptors that the drug, or similarly structured drugs, are known to target.[6] These computational methodologies demonstrated promising performance in predicting significant adverse events. These results may indicate which organ systems and adverse event categories to closely monitor during clinical trials or clinical and nonclinical data review.
Additionally, DARS is analyzing and building a database for secondary pharmacology activity submitted by the industry as part of their Investigational New Drug Application. A drug developer typically conducts in vitro target binding and functional assays for 80 to 100 biological receptors to determine potential on-target and off-target effects. However, the targets chosen for the assays and submission format are not currently standardized across the industry.
Therefore, data from these assays have been manually extracted and curated into a database to allow easier access to and analysis of these study results. Additionally, DARS is engaging in a public-private partnership with the Pistoia Alliance to determine the best methods for future regulatory submission of these studies.
Pharmacodynamic Biomarkers
Many of DARS’ current initiatives demonstrate the FDA's thinking on using pharmacodynamic (PD) biomarkers to demonstrate biosimilarity that may streamline or negate the need for comparative clinical studies (Figure).[7] This included conducting 3 clinical studies to define best practices for characterizing PD biomarkers for different classes of drugs and to develop general considerations applicable to all types of biomarkers for biological products.[8],[9],[10]

![Figure 1. The FDA perspective on developing pharmacodynamic markers for biosimilars. [Source: FDA]](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fbiosimilars%2Ff9d2b128438efe5a7827ad3b40842c6892a46931-2500x1687.png%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "Figure 1. The FDA perspective on developing pharmacodynamic markers for biosimilars. [Source: FDA]")
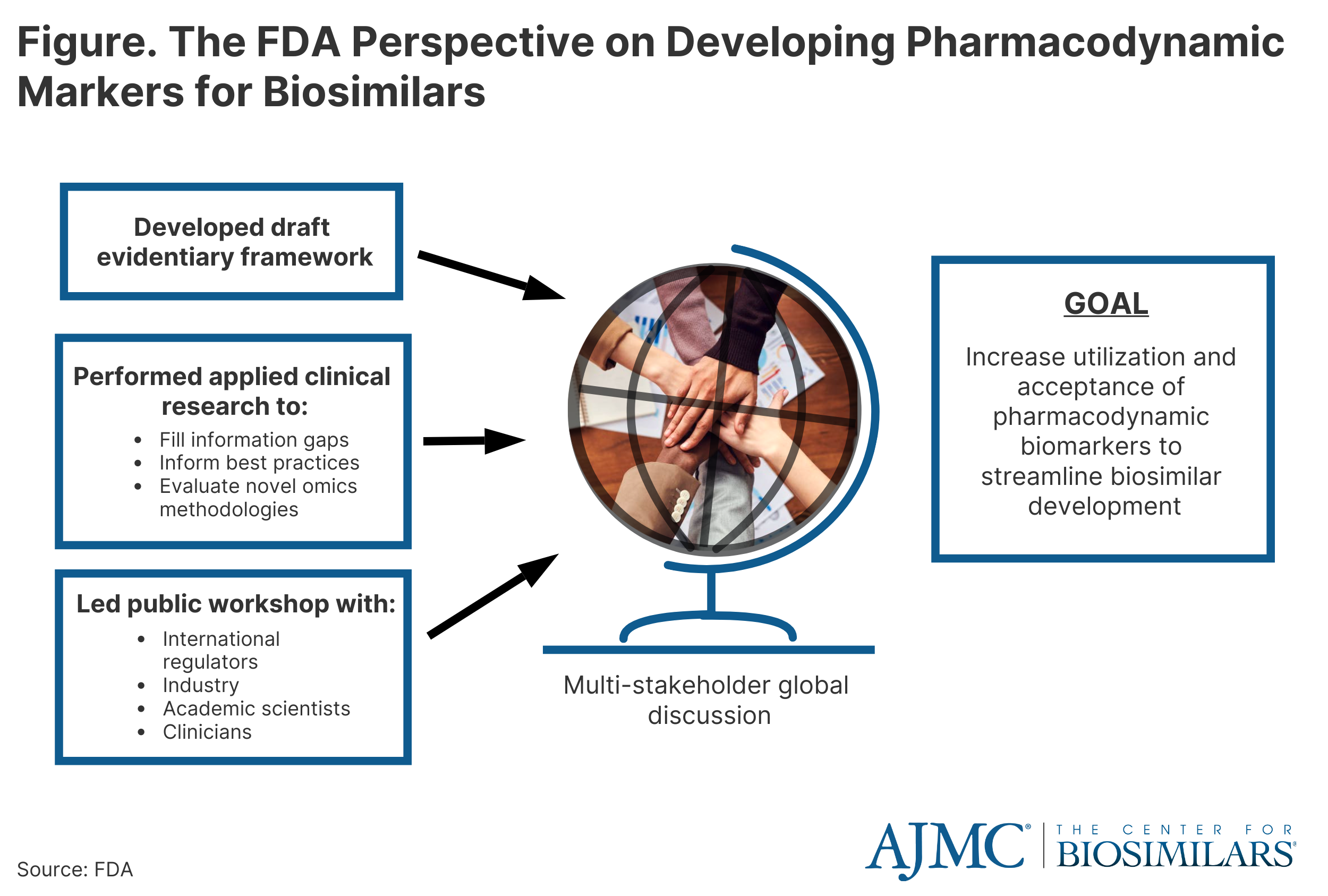
These studies included assessments to evaluate the uses of proteomic and transcriptomic analysis of human plasma to identify novel biomarkers for biosimilar development.[11] In addition, a joint FDA/Duke Margolis Workshop discussed initial findings and facilitated a broader discussion on using PD biomarkersfor biosimilar development.[12] In the journal Clinical Pharmacology and Therapeutics, additional details are reported in the January 2023 themed issue on Innovations in Biosimilars.[13]
Impurities and Extractable/Leachable Compounds
The growing use of (Q)SAR models for the safety assessment of drug products led to the formation of the DARS Computational Toxicology Consultation Service. In addition, the ICH M7(R1) international regulatory guideline recommends using (Q)SAR predictions as a state-of-the-art alternative to experimental testing to qualify a drug impurity for mutagenic potential.
Extractable/leachable compounds constitute a significant concern for biosimilars as these might alter the immunogenicity profile that arrives from manufacturing, packaging, drug delivery, and/or container closure systems. For non-mutagenic compounds with limited general toxicity data, a manual structure-activity relationship, or “read-across,” assessment based on structurally similar surrogate compounds may inform setting a permissible daily exposure limit. DARS staff review applicants’ proposed surrogates for extractable/leachable compounds and, if needed, recommend alternative surrogates based on structural, metabolic, and physicochemical considerations.
I wish to thank the FDA's DARS program for removing redundant testing that only adds cost and time to the approval of biosimilars; without such actions, making biosimilars affordable may not be possible. Incidentally, the term dars means "lesson" in Persian and Arabic.
References
[1] Chiu K, Racz R, Burkhart K, et al.(2023). New science, drug regulation, and emergent public health issues: The work of FDA’s division of applied regulatory science. Front Med.2022;9:1109541. doi: 10.3389/fmed.2022.1109541
[2] Yan H, Bhagwat B, Sanden D, Willingham A, Tan A, Knapton A, et al. Evaluation of a TGN1412 analogue using in vitro assays and two immune humanized mouse models. Toxicol Appl Pharmacol. 2019;372:57-69. doi: 10.1016/j.taap.2019.03.020
[3] Yan H, Semple K, Gonzalez C, Howard K. Bone marrow-liver-thymus (BLT) immune humanized mice as a model to predict cytokine release syndrome. Transl Res. 2019;210:43-56. doi: 10.1016/j.trsl.2019.04.007
[4] Weaver J, Zadrozny L, Gabrielson K, Semple K, Shea K, Howard KE. BLT-immune humanized mice as a model for nivolumab-induced immune-mediated adverse events: comparison of the NOG and NOG-EXL strains. Toxicol Sci. 2019;169:194-208. doi: 10.1093/toxsci/kfz045
[5] Niazi SK. Molecular biosimilarity—an AI-driven paradigm shift. Int J Mol Sci. 2022;23(18):10690. doi: 10.3390/ijms231810690
[6] Daluwatte C, Schotland P, Strauss D, Burkhart K, Racz R. Predicting potential adverse events using safety data from marketed drugs. BMC Bioinform. 2020;21:163. doi: 10.1186/s12859-020-3509-7
[7] Li J, Florian J, Campbell E, Schrieber S, et al. Advancing biosimilar development using pharmacodynamic biomarkers in clinical pharmacology studies. Clin Pharmacol Ther. 2020;107:40-2. doi: 10.1002/cpt.1653
[8] Sheikhy M, Schrieber S, Sun Q, Gershuny V, Matta M, Bai J, et al. Considerations for use of pharmacodynamic biomarkers to support biosimilar development- (I) a randomized trial with PCSK9 inhibitors. Clin Pharmacol Ther. 2022;113(2):71-79. doi: 10.1002/cpt.2769
[9] Gershuny V, Sun Q, Schrieber S, et al. Considerations for use of pharmacodynamic biomarkers to support biosimilar development - (II) a randomized trial with IL-5 antagonists. Clin Pharmacol Ther. 2022;1:80-89. doi:10.1002/cpt.2760
[10] Florian JG, Sun Q, Schrieber S, Matta M, Hazel A, Sheikhy M, et al. Considerations for use of pharmacodynamic biomarkers to support biosimilar development – (III) a randomized trial with interferon beta-1a products. Clin Pharmacol Ther. 2022:113(2):339-348. doi: 10.1002/cpt.2784
[11] Hyland P, Chekka L, Samarth D, et al. Evaluating the utility of proteomics for the identification of circulating pharmacodynamic biomarkers of IFNbeta-1a biologics. Clin Pharmacol Ther. 2022;113(1):98-107. doi: 10.1002/cpt.2778
[12] Florian JS, Schrieber S, White R, et al. Pharmacodynamic biomarkers for biosimilar development and approval: a workshop summary. Clin Pharmacol Ther. Published online November 15, 2022. Accessed January 27, 2023. doi: 10.1002/cpt.2795
[13] Strauss D, Wang Y, Florian J, Zineh I. Pharmacodynamic biomarkers evidentiary considerations for biosimilar development and approval. Clin Pharmacol Ther. 2022;113(1):55-61. doi: 10.1002/cpt.2761
Newsletter
Where clinical, regulatory, and economic perspectives converge—sign up for Center for Biosimilars® emails to get expert insights on emerging treatment paradigms, biosimilar policy, and real-world outcomes that shape patient care.









