

- Bone Health
- Immunology
- Hematology
- Respiratory
- Dermatology
- Diabetes
- Gastroenterology
- Neurology
- Oncology
- Ophthalmology
- Rare Disease
- Rheumatology
BioRationality: Entering a New Era of Affordable Biosimilar Development
Biosimilar development costs could plummet from $100 million to $5 million, enabling smaller companies to enter the market and revolutionize access to essential medicines.
The biosimilars landscape is experiencing a transformative shift. Recent regulatory developments are poised to reduce development costs from $100 million to as little as $5 million—opening doors that were previously locked to all but the largest pharmaceutical companies.
The Regulatory Revolution
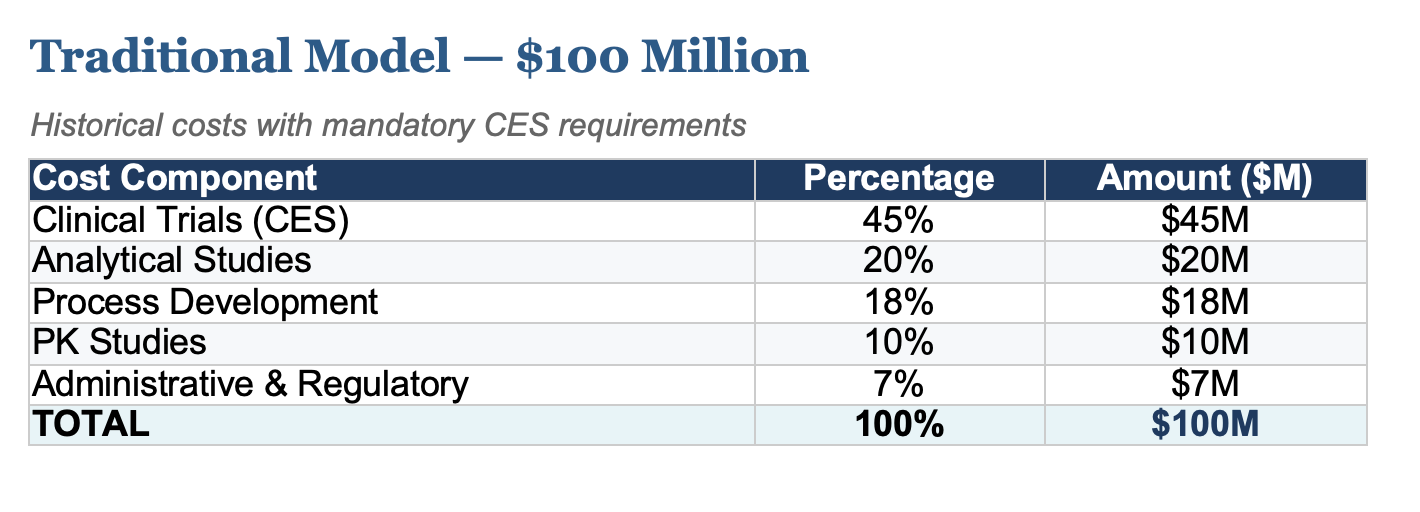
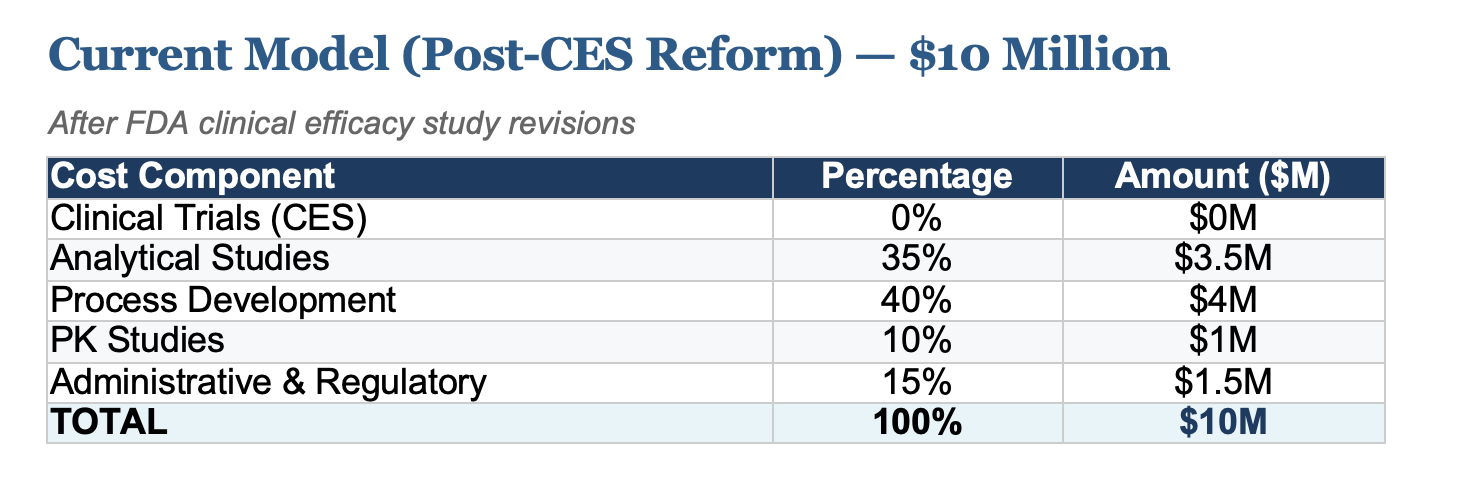
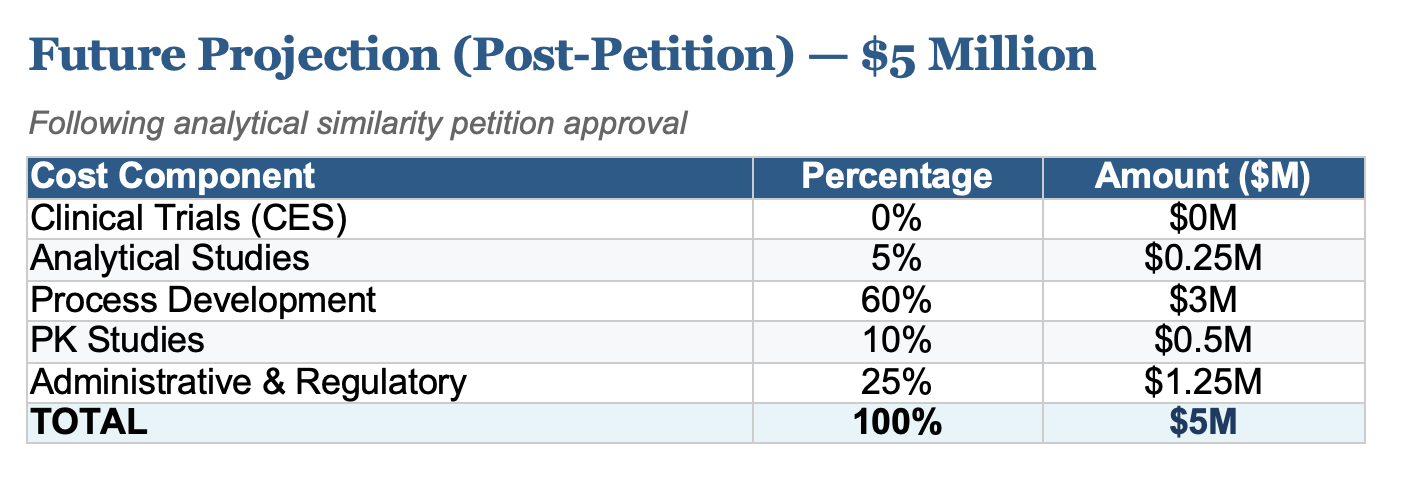
The past few months have delivered unprecedented news for the future of biosimilars. The FDA's decision to reconsider clinical efficacy studies (CES) requirements, a change I first petitioned for, has fundamentally altered the economics of biosimilar development. What once cost upwards of $100 million is now achievable at approximately $10 million, with projections showing costs could fall to just $5 million following full implementation of proposed regulatory changes.
This isn't merely incremental improvement; it's a paradigm shift. The biosimilar market, currently dominated by major pharmaceutical companies who alone could absorb the substantial development costs, is now becoming accessible to smaller, more agile companies.
The Economics Transformed
The following breakdown illustrates how development costs have evolved and where they're headed. Clinical trials, historically the largest cost driver, have been eliminated as regulatory science advances.



 — $10 Million")

 — $5 Million")
Key Cost Drivers Explained
- Clinical Trials (CES): Efficacy studies in patient populations, historically the largest expense
- Analytical Studies: Comprehensive characterization and comparability testing
- Process Development: Cell line development, upstream/downstream optimization
- PK Studies: Pharmacokinetic and pharmacodynamic bridging studies
- Administrative & Regulatory: Filing fees, documentation, quality systems
Challenges for Emerging Developers
Despite these favorable developments, smaller companies, particularly those transitioning from generic chemical drugs, face significant challenges. The biological development paradigm differs fundamentally from small molecule generics.
Critical Misconception
There is no parallel between chemical generics' strategy of importing drug substance (DS) for local conversion to drug product (DP). In biologics, the DS essentially is the DP. The FDA requires approval of both the DS supplier and the DP conversion facility, significantly expanding the regulatory workload.
Another prevalent misconception involves attempting to secure finished products from approved manufacturers and subsequently transitioning to local production. This approach fails for several reasons: companies with FDA- or European Medicines Agency-approved products rarely license to smaller entities—not merely due to cost and margin considerations, but because of post-market reporting risks. A single adverse event report from a developing country, potentially attributable to storage issues or handling, presents unacceptable liability for companies that have invested heavily in securing stringent regulatory approval.
Strategic Pathways Forward
Emerging developers face 2 primary pathways. The first involves establishing full-scale manufacturing facilities—a multi-year undertaking. Most companies, understandably, prefer the second option: engaging contract development and manufacturing organizations (CDMOs) for initial development, with plans to exercise technology transfer provisions (ICHA501) once the BLA approval is secured.
The "Make It Local, Sell It Global" Paradigm
Today, approximately 80% of the global population lacks access to these essential molecules. Even in developed markets, 90% of available biologics have no biosimilar alternatives. The market opportunity is vast and expanding.
However, CDMO engagement requires careful navigation. A critical point from my personal experience: nearly every non-US CDMO presents agreements restricting full technical documentation disclosure, offering only master file references to regulatory agencies. This arrangement fundamentally limits business expansion opportunities, as licensing out or self-manufacturing represents the most sustainable business model.
The Path Forward
When engaging a CDMO, the first and most critical question should be: "Will I receive 100% of the technical documentation?" Any response qualified with "if" or "but" should prompt immediate reconsideration. Many CDMOs can and will comply with full transparency requirements.
From $100M → $10M → $5M: The democratization of biosimilar development
A New Chapter Begins
I am now more enthused and optimistic than ever that many smaller companies will enter the biosimilar business. Within five years, these essential medicines will be as affordable as generic drugs—the ultimate goal that has driven this work from the beginning. The expansion from just 13 molecules to over 200 available biosimilars is not just possible; it is now within reach.
Newsletter
Where clinical, regulatory, and economic perspectives converge—sign up for Center for Biosimilars® emails to get expert insights on emerging treatment paradigms, biosimilar policy, and real-world outcomes that shape patient care.









