

- Bone Health
- Immunology
- Hematology
- Respiratory
- Dermatology
- Diabetes
- Gastroenterology
- Neurology
- Oncology
- Ophthalmology
- Rare Disease
- Rheumatology
Opinion: A Rational Approach to Establish Biosimilarity
Sarfaraz K. Niazi, PhD, a member of The Center for Biosimilars® Advisory Board, reports on his citizen petition to the FDA to rationalize testing of biosimilars.
Section 7002 of the Biologics Price Competition and Innovation Act, which details the approval pathway for biosimilar biological products, has come a long way with the issuance of several FDA guidance documents—some revised and 1 withdrawn. Developers and the FDA have collaborated on rewriting recommendations for establishing biosimilarity that will support the Section 7002 mandate to enable faster development of biosimilars, benefiting American patients. This column is abstracted from my citizen petition to the FDA on the same topic.

Although all FDA guidelines include a disclaimer that the guidance provided is suggestive and neither the developer nor the FDA is bound by it, this disclaimer is universally ignored, and the developers try to comply with the listed recommendations. I recommend that the FDA look at the suggestions made here and create a new guideline that will be rational and less demanding on developers without compromising the safety and efficacy of biosimilars.
In submitting my thesis, I am introducing a few new terms. Legacy attributes are the critical quality attributes (CQAs) for which release specifications have been well-established. There is no need to test these attributes side by side with the reference product. Nonlegacy attributes are established based on the testing of the reference product, but not necessarily in a comparative mode. Qualification testing is intended to verify that the active molecule sufficiently matches the reference product, and all of this testing is conducted on a smaller number of batches.
Creating a rational approach requires examining all CQAs based on the relevance and appropriateness of testing. The CQAs are divided into product-related and process-related attributes; the former require comparative testing with the reference product and the latter only if a legacy range of variability cannot be established. This concept is presented in the Figure.


Click to enlarge.
CQAs comprise the properties that can affect the safety and safety of the product. How these attributes are tested should be segregated by product attributes associated with the expression system and process attributes associated with the manufacturing process. The product attributes are the least variable, as they should be, and establish the suitability of the product for further development. These attributes should not be compared with legacy values such as the structure reported in the literature; the comparisons must be made in side-by-side testing with the reference product. Because the product-related attributes have narrow acceptance criteria, only 3 lots of biosimilar candidates and reference products would suffice for testing. Forced degradation studies can be considered as both product and process-related attributes as they tell much about the inter- and intramolecular bindings determined by the primary, secondary, and tertiary structures. Unfortunately, the World Health Organization does not recognize the value of forced degradation or accelerated stability testing in a comparative mode, missing out on a critical method of evaluating molecular structure.
The molecular structure is further confirmed through a biological lens that can comprise in vitro pharmacology, nonclinical pharmacology (pharmacokinetic [PK] studies in animals). Animal PK studies help support structural similarity through the eyes of a biological system to view the molecules, albeit the disposition profiles are different in humans. Although such studies are required for antibody testing in larger species, these should be required for all products.
The most important test is the human PK/pharmacodynamic (PD) study based on how the body sees the molecules and how the molecules see the body. Novel study protocols can combine the PK/PD and immunogenicity testing in a single study that can be made more powerful by narrowing down the inclusion/exclusion criteria, because the purpose of these studies is to compare, not characterize, the profiles. One such type of study can be a 2-periods, parallel, 2-dose study.
Process-Related Testing
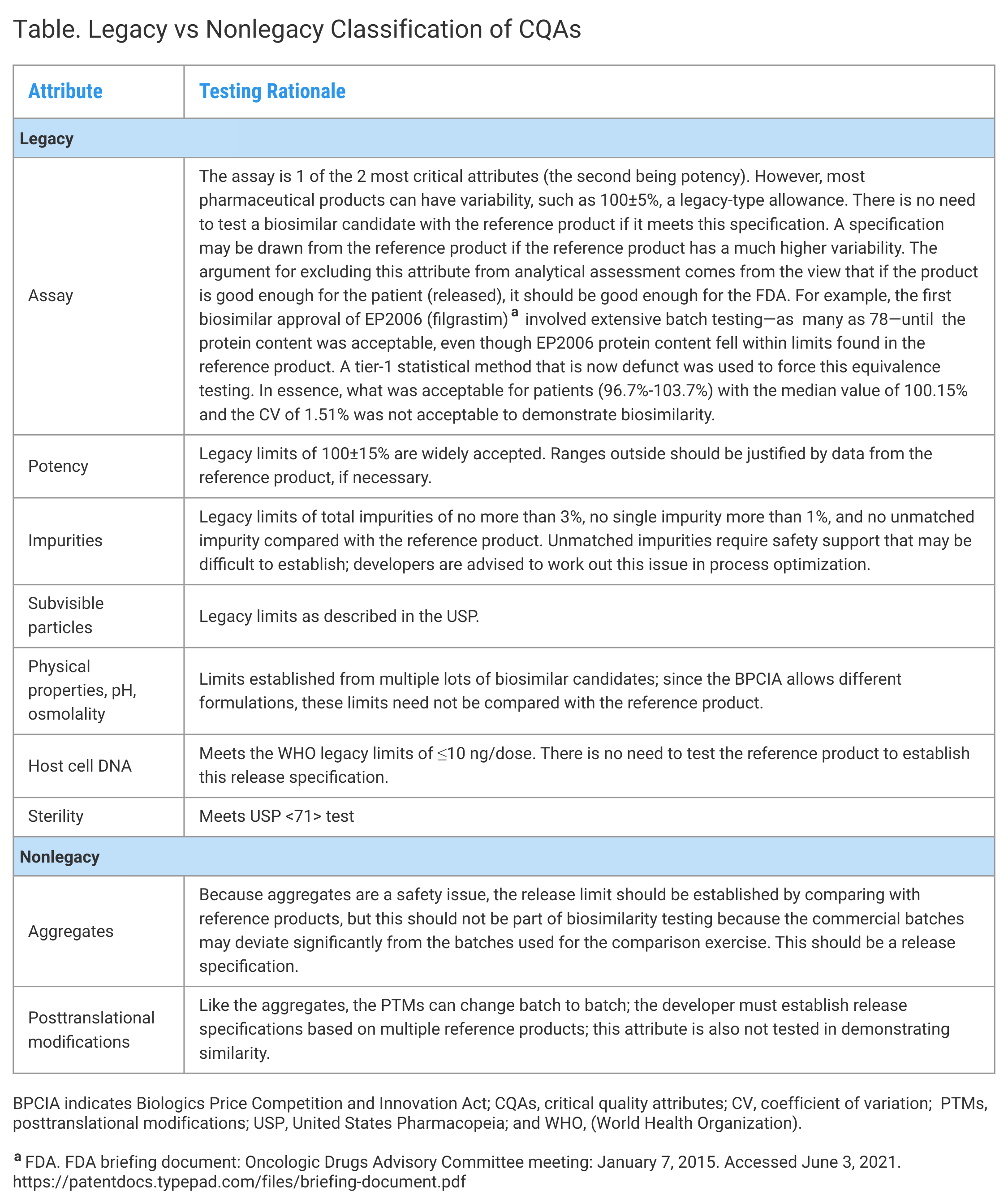
Although the expression system determines the product's structure, the rest of the variations result from differences and variabilities in the process, from upstream to downstream to the formulation. These CQAs cannot be 1-time testing and should be included in the release specifications. The CQAs can be classified into 2 categories: 1 where the release limits are well-known and well-established, and the other where the limits must be established based on the ranges obtained from the reference product. This concept of similarity establishment is efficient and based on scientific principles, and it reduces the burden of testing substantially (Table).


Click to enlarge.
Redundant Studies
Animal toxicology studies are not needed, because the molecule's safety has been established, and any minor differences in the structure or posttranslational modificationprofile will not be detected because the testing is conducted at the higher end of the linear range of toxicity. In most cases, the animals do not have similar receptors, which makes these studies least relevant. This same argument holds for any PD and immunogenicity testing in animals. There is also little correlation with the human dose to make such studies redundant. The European Union and FDA have approved more than 100 products, and none of the products failed in animal toxicology testing because they cannot.
Clinical efficacy and safety studies comprise the most significant time and cost hurdles in the development of biosimilars, even though these studies do not add any assurance to the safety and efficacy evaluation. Hundreds of clinical efficacy and safety studies reported were conducted without the European Union or the FDA establishing that there is any “residual uncertainty” primarily to support the marketing of these products. However, if a biosimilar product fails to meet any of the studies stated above, particularly the clinical pharmacology, the FDA does not allow testing in patients because these studies can never resolve any “residual uncertainty” for the following reasons:
- A single study in 1 of the several possible indications does not resolve uncertainty even where the mechanisms of action are cross-validated.
- Efficacy studies require an arbitrary selection of differences in efficacy based on a clinical judgment that can vary widely.
- A noninferiority study may allow a product with higher efficacy and, therefore, a possible higher safety risk.
- Clinical markers are often unreliable and, in some populations like cancer patients, highly variable.
- Many studies lose patients in critical care and rarely find naïve patients to reduce the variability and increase test power.
- The statistical model of demonstrating that 2 products are not different is more complex than showing the efficacy of a product against a placebo.
None of the biosimilars tested for safety or efficacy failed the test. The clinical safety and efficacy are already subject to postmarket drug and biologic safety evaluations as required by the FDA.
Summary
In evaluating biosimilars, the FDA has created precise and scientifically significant vocabulary terms such as “no clinically meaningful difference” and “residual uncertainty” that are relevant and represent a creative approach to ensuring the safety of biosimilars. However, certain practices are needed to enable faster approval of biosimilars.
The following is a summary of my recommendations to the FDA:
- Reduce comparative similarity assessment to relevant attributes.
- Eliminate animal toxicology studies.
- Add animal pharmacology studies
- Allow inclusion criteria in clinical pharmacology studies to reduce intersubject variability
- Define “residual uncertainty” for every product evaluated and reject those where a resolution cannot be made.
- Rely on postmarket evaluation for safety and efficacy instead of limited testing that cannot support removing any residual uncertainty.
Newsletter
Where clinical, regulatory, and economic perspectives converge—sign up for Center for Biosimilars® emails to get expert insights on emerging treatment paradigms, biosimilar policy, and real-world outcomes that shape patient care.









