

- Bone Health
- Immunology
- Hematology
- Respiratory
- Dermatology
- Diabetes
- Gastroenterology
- Neurology
- Oncology
- Ophthalmology
- Rare Disease
- Rheumatology
The Pandemic's Multilevel Effect on Patient Access
Coronavirus disease 2019 altered the workflow in all areas of the health care and life sciences industries, and the FDA was not exempt from pandemic-induced upheaval.
Coronavirus disease 2019 (COVID-19) altered the workflow in all areas of the health care and life sciences industries. The FDA was not exempt from pandemic-induced upheaval. Meetings of advisory committees, which provide the FDA with expert advice on scientific, technical, and policy matters, were often postponed, and although drugs were still reviewed and approved, significant delays occurred.
From the start of the United States’ response to the COVID-19 pandemic in March 2020 until late summer, 2 biosimilars were approved: Hulio (adalimumab) and Nyvepria (pegfilgrastim). This represented not only an improvement in patient access and affordability of life-changing medications, but also a slowdown in biosimilar approvals.
In previous years, biosimilar approvals had steadily increased. As shown in the figure, the projected trend suggested approximately 12 biosimilar approvals for 2020 and 14 for 2021.


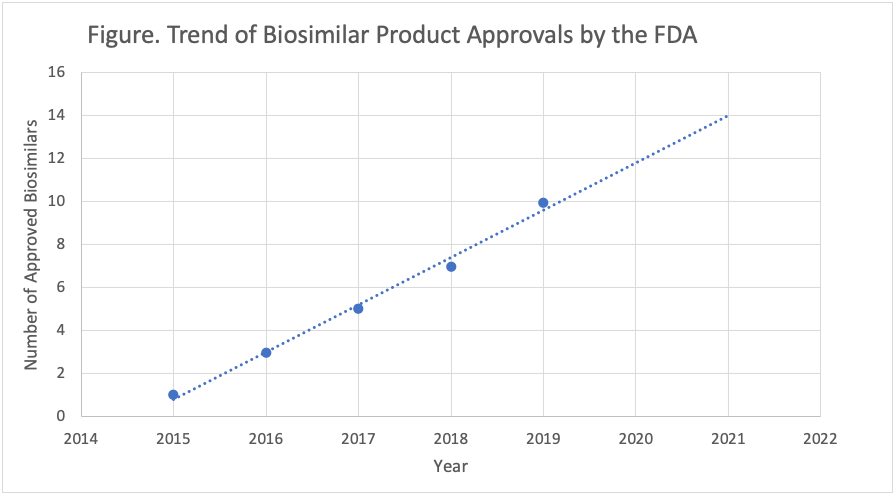
The trend line for biosimilar approvals suggested 2020 would be another strong year for the emergence of these lower-cost therapies; however, just 2 had been approved by the midyear point.
Although other factors came into play, such as patent expirations and product development, it appeared unlikely that the total of 12 biosimilar approvals would be achieved in the final quarter of 2020, and there was even potential for this slower pace to extend into 2021.
There were other reasons why patient access did not improve as expected. Typically, once manufacturers receive biosimilar approval, many face lawsuits that delay products from reaching the market promptly. These court cases often are purposely filed to interfere with market access to a new product. In some cases, “pay-for-delay” agreements postpone market entry of a biosimilar for many years.
The COVID-19 crisis extended these delays because court systems temporarily closed to prevent spread of the virus and reopenings were late or occurred only in stages. Well into 2020, virus hot spots continued to stall judiciary system operations, adding to the time needed to achieve market access for these new therapy options.
In addition to regulatory and judicial barriers, the overwhelming workflow within health systems also played a role in limiting access to new therapies. When health care facilities are in crisis mode, many components of the reimbursement and education processes, each of which is vital for biosimilar uptake, are hindered.
Pharmacy and therapeutics (P&T) committees are necessary for ensuring new therapies will be included in the health care facility formulary. From the start of COVID-19, P&T meetings were often conducted virtually or canceled. Many P&T efforts centered on evaluating emerging therapies for COVID-19 treatment, leaving little time for consideration of other formulary additions. With so much focus on COVID-19, little time was allocated to much else.
Within hospitals, the same was true regarding in-service education for physicians. With a need to educate physicians on current protocol, diagnostics, and treatment surrounding COVID-19, there was not much time left to educate on biosimilar therapies and novel agents.
“Lunch and learn” programs were likely on hiatus during the pandemic due to social distancing requirements, understaffing, and a strain on funding. Gathering professionals into a conference room could not be prioritized over keeping everyone safe.
Typically, many facilities partner with manufacturers to supply marketing materials and food to lunch and learns, and a clinical staff member presents on the topic at hand. As manufacturer field teams were grounded, less information reached the health care providers. Most nonessential visits were prohibited to prevent unnecessary risk of virus spread. These grounded professionals included sales representatives who provide information about new products, field reimbursement specialists who assist in getting products approved by the health plans, and medical science liaisons who can discuss new products on a higher level. These professionals were not as physically active as the pandemic unfolded and virtual methods of interaction were implemented.
If new therapies, including biosimilars, were not approved by the FDA, litigated promptly in the courts, cleared for formulary placement, or explained to physicians, it is unlikely patients were learning much about them. Especially when it comes to biosimilars, patients need to be aware of their potential options and the associated benefit of cost savings. With COVID-19, the nation entered a time of financial instability and loss of health care coverage, and these new therapy options became even more important for safe and affordable health care.
Newsletter
Where clinical, regulatory, and economic perspectives converge—sign up for Center for Biosimilars® emails to get expert insights on emerging treatment paradigms, biosimilar policy, and real-world outcomes that shape patient care.









